Marell, D. J.; Emond, S. J.; Kulshrestha, A.; Hoye, T. R. J. Org. Chem. 2014, 79, 753–758.
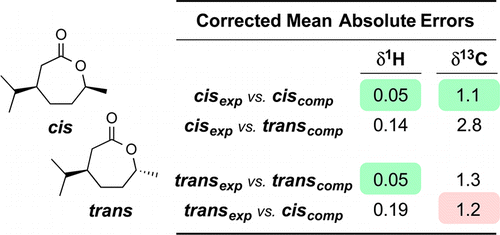
We report an NMR chemical shift study of conformationally challenging seven-membered lactones (1–11); computed and experimental data sets are compared. The computations involved full conformational analysis of each lactone, Boltzmann-weighted averaging of the chemical shifts across all conformers, and linear correction of the computed chemical shifts. DFT geometry optimizations [M06-2X/6-31+G(d,p)] and GIAO NMR chemical shift calculations [B3LYP/6-311+G(2d,p)] provided the computed chemical shifts. The corrected mean absolute error (CMAE), the average of the differences between the computed and experimental chemical shifts for each of the 11 lactones, is encouragingly small (0.02–0.08 ppm for 1H or 0.8–2.2 ppm for 13C). Three pairs of cis versus trans diastereomeric lactones were used to assess the ability of the method to distinguish between stereoisomers. The experimental shifts were compared with the computed shifts for each of the two possible isomers. We introduce the use of a “match ratio”—the ratio of the larger CMAE (worse fit) to the smaller CMAE (better fit). A greater match ratio value indicates better distinguishing ability. The match ratios are larger for proton data [2.4–4.0 (av = 3.2)] than for carbon [1.1–2.3 (av = 1.6)], indicating that the former provide a better basis for discriminating these diastereomers.