Willoughby, P. H.; Jansma, M. J.; Hoye, T. R. Nat. Protoc. 2014, 9, 643–660.
Addendum: Nat. Protoc. 2020, 15, 2277. (DOI: 10.1038/s41596-020-0293-9)
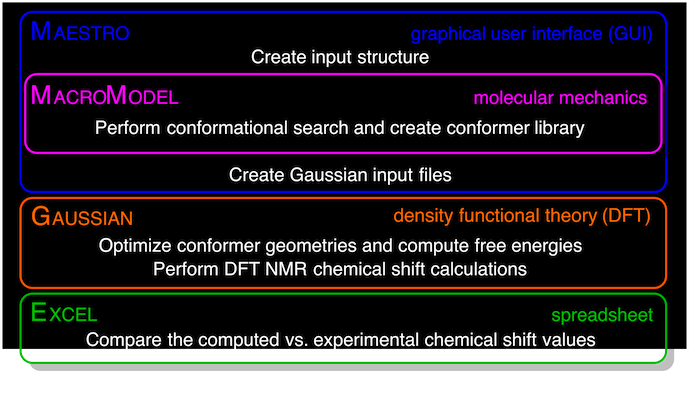
This protocol is intended to provide chemists who discover or make new organic compounds with a valuable tool for validating the structural assignments of those new chemical entities. Experimental 1H and/or 13C NMR spectral data and its proper interpretation for the compound of interest is required as a starting point. The approach involves the following steps: (i) using molecular mechanics calculations (with, e.g., MacroModel) to generate a library of conformers; (ii) using density functional theory (DFT) calculations (with, e.g., Gaussian 09) to determine optimal geometry, free energies and chemical shifts for each conformer; (iii) determining Boltzmann-weighted proton and carbon chemical shifts; and (iv) comparing the computed chemical shifts for two or more candidate structures with experimental data to determine the best fit. For a typical structure assignment of a small organic molecule (e.g., fewer than ∼10 non-H atoms or up to ∼180 a.m.u. and ∼20 conformers), this protocol can be completed in ∼2 h of active effort over a 2-d period; for more complex molecules (e.g., fewer than ∼30 non-H atoms or up to ∼500 a.m.u. and ∼50 conformers), the protocol requires ∼3–6 h of active effort over a 2-week period. To demonstrate the method, we have chosen the analysis of the cis- versus the trans-diastereoisomers of 3-methylcyclohexanol (1-cis versus 1-trans). The protocol is written in a manner that makes the computation of chemical shifts tractable for chemists who may otherwise have only rudimentary computational experience.