Wang, T.; Niu, D.; Hoye, T. R. J. Am. Chem. Soc. 2016, 138, 7832–7835.
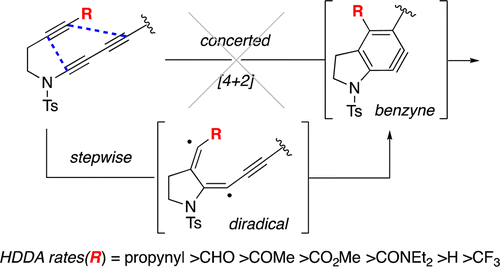
We report here experiments showing that the hexadehydro-Diels–Alder (HDDA) cycloisomerization reaction proceeds in a stepwise manner—i.e., via a diradical intermediate. Judicious use of substituent effects was decisive. We prepared (i) a series of triyne HDDA substrates that differed only in the R group present on the remote terminus of the diynophilic alkyne and (ii) an analogous series of dienophilic alkynes (n-C7H15COC≡CR) for use in classical Diels–Alder (DA) reactions (with 1,3-cyclopentadiene). The R groups were CF3, CHO, COMe/Et, CO2Me, CONMe2/Et2, H, and 1-propynyl. The relative rates of both the HDDA cyclization reactions and the simple DA cycloadditions were measured. The reactivity trends revealed a dramatic difference in the behaviors of the CF3 (slowest HDDA and nearly fastest DA) and 1-propynyl (fastest HDDA and slowest DA) containing members of each series. These differences can be explained by invoking radical-stabilizing energies rather than electron-withdrawing effects as the dominating feature of the HDDA reaction.